Beyond Targets: Building Safer Drugs with Integrated AI, Toxicology, and Experiment Loops

How the right orchestration of AI, in silico tool and experimentation can optimize drug development.
Introduction
Many multibillion-dollar tech companies have been founded, led, and scaled by small teams. From the traditional perspective of drug discovery, moving this concept from tech to life sciences might seem impractical. Yet the explosion of computational tools and AI agents is quickly transforming what once appeared unfeasible into a new reality. Today, no matter what your original training, the tools available make it possible to analyze, learn, experiment, and understand fields that were only a few years ago inaccessible without formal study and deep domain expertise. This opens the door to lean, agile organizations that require less funding and operate on more aggressive timelines to reach critical value-inflection points.
Importantly, it also gives non-scientist experts the ability to access knowledge easily that can be directly useful in their day-to-day work. Picture an investment analyst who, with an accessible bio-platform, could gain deep technical insight into the profile of a therapeutic agent and immediately form a clear understanding of that company's investment prospects.
The Big Picture: Why Profiling Molecules Matters as Much as Target Choice
Accessible AI and in silico tools now enable small teams, including those without formal chemistry training, to gain critical insights into potential toxicology flags for small molecules of therapeutic interest. While many powerful tools are freely available, they are scattered across different applications, websites, and software packages, making analyses that should be routine more complicated and less straightforward than necessary. (Fig. 1)
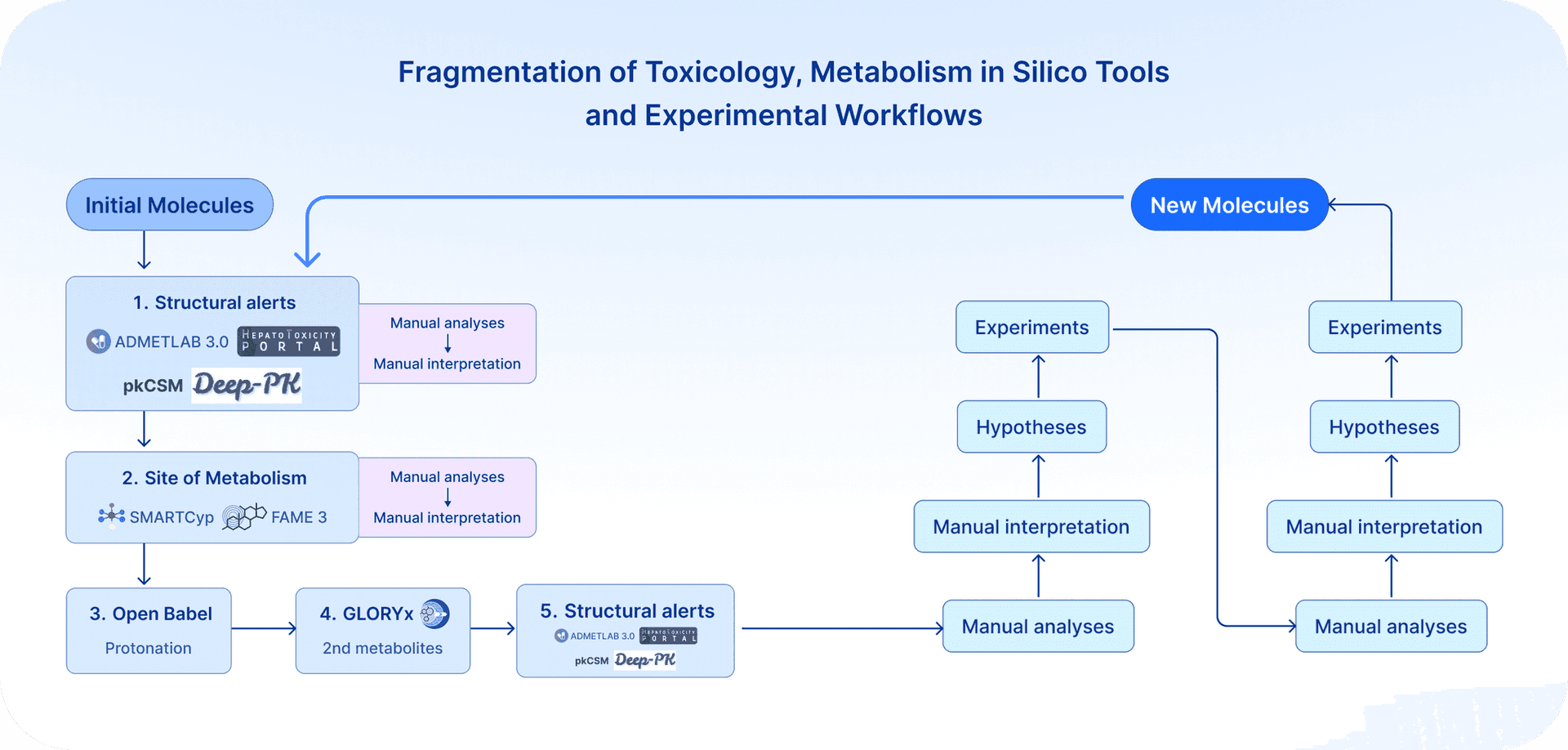
Using USP1 inhibitors as a case study, we applied a suite of in silico tools to understand why TNG-348 displayed liver toxicity in clinical trials while KSQ-4279 did not. Although these remain predictive assessments and integration with experimental data is essential, it is nonetheless striking how much insight can be gained without deep chemistry expertise into the mechanistic differences that can potentially explain divergent clinical outcomes.
Here is the list of tools used; some are web-based and others are software applications: Toxtree, SMARTCyp, FAME3, GLORYx, ADMET 3.0 / pkCSM, Deep-PK, KOBIC / HTP platform, ADMETlab 3.0, Open Babel (see box for description)
Box description
- Toxtree – software application for structural hazard alerts, Cramer's rules, protein/DNA binding, and CYP metabolism trees
- Open Babel – software application for file format conversion, molecular preprocessing, and descriptor generation
- SMARTCyp – web-based predictor of CYP450 sites of metabolism, widely used for isoforms like CYP3A4, CYP2D6, and CYP2C9
- FAME3 – web-based predictor of metabolic hot spots with probabilities and applicability domain scoring
- GLORYx – web-based platform for secondary metabolite generation and mechanistic biotransformation prediction
- ADMET 3.0 / pkCSM – web-based predictors of pharmacokinetics, clearance, free fraction, and transporter liabilities such as BCRP, OATP, and BSEP
- Deep-PK – web-based AI platform for ADMET profiling, hepatotoxicity models, and stress reporters such as AhR, ARE/Nrf2, ATAD5, and p53.
- KOBIC / HTP platform – web-based interface for structural toxicity alerts and chemical safety screening
- ADMETlab 3.0 – web-based tool for integrated ADMET and toxicity predictions
Imagine running the whole stack in one place with a single click: structural alerts, sites of metabolism, secondary metabolite generation, PK and transport predictions, and cellular stress reporters. Imagine being able to streamline in silico predictive tools with experimental data and iterate everything in a simple, accessible loop. That is the kind of integrated platform we believe would be transformative for modern drug discovery. That is our goal, that is our vision, and not just for chemistry. (Fig. 2)
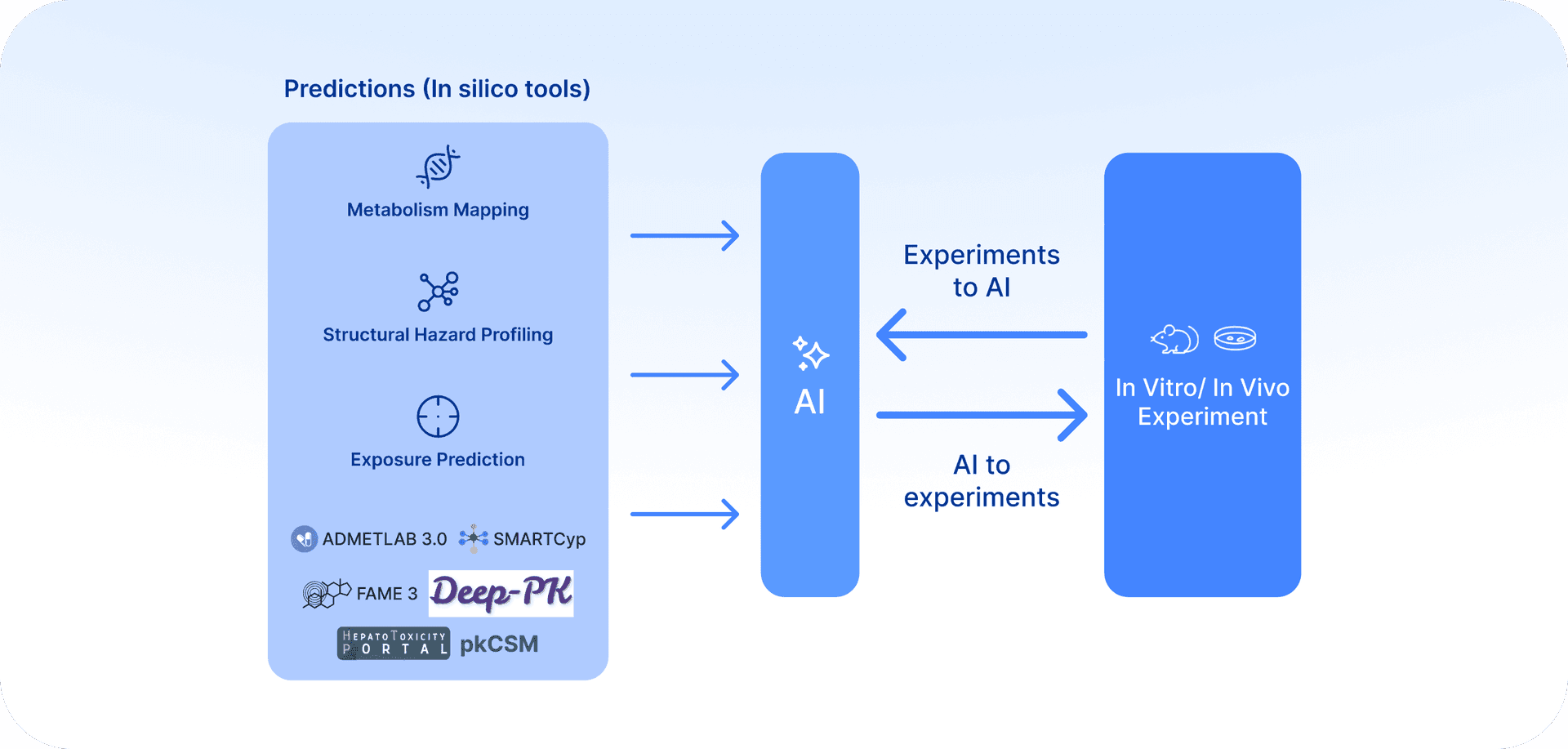
In our previous article, we argued that multidimensional data integration can de-risk target identification. But even the best target choice will falter if the chemotype chosen to drug it encodes hidden liabilities. Roughly one-third of clinical failures stem from safety problems, many driven not by biology but by scaffold-embedded risks such as toxicophores, metabolic soft spots, and transporter interactions.
Lessons from Comparative Case Studies: KSQ-4279 vs TNG-348
Biology Meets Chemistry
To illustrate how chemotype choice can determine clinical fate, we examined two related compounds: KSQ-4279, which advanced with a tolerable profile, and TNG-348, which was halted in Phase I for liver toxicity. But drug metabolism does not stop at the parent molecule. Secondary metabolites are formed when initial metabolites undergo further enzymatic transformation within the body, and these downstream products can sometimes be even more reactive and toxic than the original compound. In addition to profiling the parent molecules, we also derived their predicted secondary metabolites in silico and assessed these for metabolic and toxicological liabilities.
Despite similar scaffolds, a layered in silico analysis revealed consistent differences:
Metabolism: When a compound is highly metabolizable, the body breaks it down quickly and through many routes. This raises the chance of generating reactive intermediates. Both compounds are readily metabolized, but TNG-348 displays a broader set of high-probability metabolic hot spots. Among these is a tertiary amine position prone to oxidation into reactive iminium intermediates, which can bind to proteins or DNA and drive liver toxicity. This broader footprint extends into TNG-348's secondary metabolites, which also show more routes prone to bioactivation.
Structural hazards: These are chemical features that can generate reactive species or bind to proteins and DNA. Both KSQ-4279 and TNG-348 show baseline alerts for hepatotoxicity, so structural flags alone does not explain why TNG-348 showed a clinical hepatotoxicity signal while KSQ-4279 did not. Interestingly, TNG's secondary metabolites consistently carry a heavier burden, with more DNA-binding and protein-binding liabilities. Several TNG metabolites sit on bioactivation-prone routes, increasing the likelihood of forming covalently reactive intermediates that can bind to proteins or DNA, reinforcing their higher hazard potential.
Exposure: How a compound is cleared and transported determines how long it stays inside liver cells, which is critical for toxicity risk. Predictions showed that TNG-348 and its metabolites have reduced BCRP efflux and modestly slower clearance, favoring greater intracellular accumulation in hepatocytes compared with KSQ-4279.
CYP burden: Cytochrome P450 enzymes (CYPs) are responsible for drug metabolism. High substrate load or inhibition of key isoforms can create metabolic stress and raise the risk of drug–drug interactions. TNG-348 is predicted to be a stronger substrate for CYP1A2 and CYP3A4 and an inhibitor of CYP2D6, giving it a heavier CYP burden than KSQ-4279. The metabolite-level analyses strengthen this view, showing greater CYP substrate turnover and inhibition liabilities across TNG's series, indicating higher metabolic stress within hepatocytes.
Stress signatures: Cellular stress reporters capture how compounds activate protective or damage-response pathways, which are often early signals of toxicity. TNG-348 and its secondary metabolites are predicted to drive stronger activation of AhR (xenobiotic sensing), ARE/Nrf2 (oxidative stress), and ATAD5 (replication stress). The Liver-Injury II model also shifts the entire TNG profile upward, reinforcing its higher hepatotoxic liability compared with KSQ-4279.
Taken together, these orthogonal signals suggest why two closely related scaffolds can produce opposite clinical outcomes. In silico tools provide valuable early insight, but experimental data remain essential, ultimately driving decision-making and scaffold selection.
By any means, we want to stress that these are extremely complex decisions involving many moving pieces, and it is extremely difficult to fully predict clinical outcomes or drawbacks. Therefore, we truly believe that a unified platform seamlessly integrating in silico predictions with experimental datasets spanning multiple layers of biology, chemistry, and clinical insights would be key to optimizing and accelerating drug development.
Box. What the KSQ-4279 vs TNG-348 Case Teaches Us
Chemotype selection can be as decisive as target choice. Even with similar target and biological rationale, scaffold-embedded liabilities can determine whether a program progresses or fails.
A Framework to De-Risk Chemotype Selection
Building on our framework for target ID, a complementary structure emerges for chemotype evaluation:
Structural Hazard Profiling (Toxtree, ADMETlab 3.0, and KOBIC) Flag toxicophores, electrophiles, and reactive metabolite precursors early.
Metabolism & Bioactivation Mapping (SMARTCyp, FAME3, Toxtree and GLORYx) Overlay SMARTCyp/FAME3 soft spots with Toxtree/GLORYx bioactivation alerts to pinpoint risky transformations. GLORYx can also generate predicted secondary metabolites for deeper liability assessment.
Exposure & Stress Integration (ADMET 3.0, pkCSM, Deep-PK, Open Babel for preprocessing) Combine pharmacokinetic and transporter predictions from ADMET 3.0 and pkCSM with Deep-PK stress reporters (AhR, ARE, ATAD5, p53) to judge whether liabilities will manifest at clinically relevant exposures.
This integrative workflow moves safety evaluation upstream, enabling together with experimental data early scaffold triage, rational substitution, or targeted mitigation strategies.
Limitations and Practical Considerations
These findings are derived entirely from predictive in silico models using freely available, non-commercial tools. While they capture mechanistic liabilities that potentially align with observed clinical outcomes, their specificity remains imperfect. No single prediction is decisive; risk separation emerges only when orthogonal signals converge across metabolism, structural hazards, clearance, transport, and stress pathways, and are further confirmed by experimental validation.
Accordingly, these results should be viewed as early triage tools that guide prioritization, scaffold substitution, and experiment planning. Critical findings must be validated with wet-lab assays such as glutathione trapping, hepatocyte covalent binding, transporter efflux studies, and CYP-mediated metabolism profiling. Integration with the prospective clinical context, including risk–benefit balance, therapeutic window, and intended indication, remains essential before making any decision to advance or terminate a program. (Fig. 3)
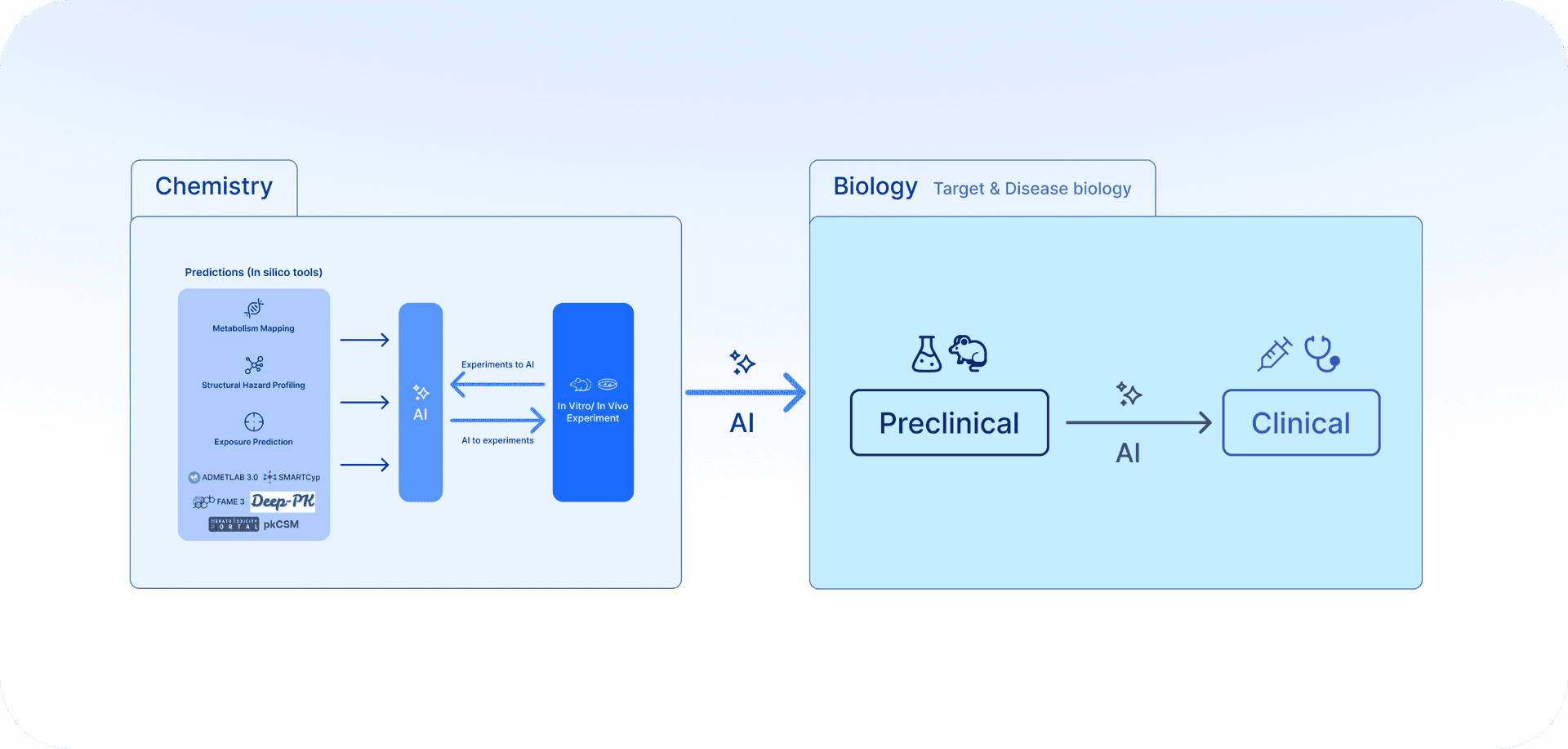
From In Silico to Experiment: Why Integration Matters
Despite all the progress that LLMs are achieving, a modern drug discovery platform cannot be only an AI model interface. To be credible and effective, it must bridge predictions and experiments.
In practice, that means creating a platform that automates and fully integrates in silico insights to design targeted assays (e.g., GSH-trapping for reactive intermediates, hepatocyte covalent binding, transporter efflux), runs them either internally or through CRO partnerships, and then feeds the results back into the computational workflow, an efficient system that facilitates key decisions and smooth progress.
Only through an easily accessible and user-friendly platform this automated loop, prediction → experiment → reintegration, can enable teams to move from early hypotheses to confident, unified conclusions in a very efficient way. The true value is not AI in isolation, but an AI-enabled ecosystem where mechanistic predictions and empirical data reinforce each other.
Closing Thoughts
Target and chemotype choices are inseparable: one defines what biology to drug, the other defines whether it can be safely drugged. The KSQ vs TNG case illustrates how scaffold-level liabilities can undo potentially compelling biology and drug discovery programs.
Together, target-level integration (Part I of this series of articles) and chemotype-level integration (Part II) outline a broader paradigm for lean drug discovery: building decision frameworks that span from biology to chemistry. With modern computational platforms that connect seamlessly to CROs and experimental pipelines, even small teams can anticipate risks earlier, reduce attrition, and accelerate the path from concept to patient benefit.
References
1. Patlewicz, G., Jeliazkova, N., Safford, R. J., Worth, A. P., & Aleksiev, B. (2008). An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR and QSAR in Environmental Research, 19(5–6), 495–524.
2. Fu L., Shi S., Yi J., Wang N., He Y., Wu Z., Peng J., Deng Y., Wang W., Wu C., Lyu A., Zeng X., Zhao W., Hou T., Cao D. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Research, 2024; Volume 52, Issue W1, Pages W422–W431.
3. Rydberg, P., Gloriam, D. E., Zaretzki, J., Breneman, C., & Olsen, L. (2010). SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Medicinal Chemistry Letters, 1(3), 96–100.
4. Kirchmair, J., Göller, A. H., Lang, D., Kunze, J., Testa, B., Wilson, I. D., Glen, R. C., & Schneider, G. (2013). Predicting drug metabolism: Experiment and/or computation? Nature Reviews Drug Discovery, 12(8), 625–640.
5. Pires, D. E. V., Blundell, T. L., & Ascher, D. B. (2015). pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of Medicinal Chemistry, 58(9), 4066–4072.
6. O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. Journal of Cheminformatics, 3(1), 33.
7. de Bruyn Kops C, Šícho M, Mazzolari A, Kirchmair J. GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chemical Research in Toxicology. 2020;33(10):2713–2727.
8. Han, J., Zhung, W., Jang, I., Lee, J., Kang, M. J., Lee, T. D., Kwack, S. J., Kim, K.-B., Hwang, D., Lee, B., Kim, H. S., Kim, W. Y., & Lee, S. (2025). KOBIC HTP: A web-based high-throughput toxicity prediction platform for chemical risk assessment. Journal of Cheminformatics, 17, 104.